Frequently Asked Questions about Medical Devices
-
Is my product a Medical Device?
'Medical device’ means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations, and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
The following products shall also be deemed to be medical devices:
- devices for the control or support of conception;
- products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
See Article 2 of MDR.
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745https://health.ec.europa.eu/system/files/2023-09/md_borderline_manual_en.pdf
https://health.ec.europa.eu/system/files/2023-06/mdcg_2022-5_en.pdf
Note: All Veterinary products are out.
-
What are the risk classes of the medical devices?
Class I – low risk.
Class I medical devices in the EU have the lowest perceived risk. In many cases, the manufacturer can self-certify Class I devices without the involvement of a notified body.This risk class includes products like stethoscopes, bandages, or glasses.
However, there are three more sub-classifications within Class I medical devices that have a slightly higher perceived risk and do require the involvement of a notified body before the manufacturer can affix the CE marking.
The sub-classifications for Class I devices are as follows (it requires a NoBo):
Class Is: The medical device must be presented sterile.
Class Im: The medical device has a measuring feature.
Class Ir: The medical device is a reusable surgical instrument.Class IIa – medium risk (it requires a NoBo): Examples of this risk class include catheters, hearing aids, or short-term contact lenses.
Class IIb – medium risk. (it requires a NoBo): This risk class includes devices like incubators, insulin pens, long-term contact lenses, and ventilators.
Class III – high risk. (it requires a NoBo): This risk class includes devices like pacemakers, prosthetic heart valves, surgical mesh, breast implants, and other devices that require permanent monitoring throughout their lifetimes.
-
What is MDR?
MDR stands for Medical Device Regulation – Regulation EU 2017/745, with corrigendum and amendments to replace the previous directives: Directive 90/385/EEC on active implantable devices and directive 93/42/EEC on medical devices.
The medical device regulation aims to ensure the safety, quality, and performance of medical devices, as well as to protect the health and rights of patients and users. -
What is a legacy device?
Legacy devices under the EU MDR and IVDR are those devices allowed to be placed on the market after the date of application of the corresponding regulation (i.e. 26 May 2021 for the EU MDR and 26 May 2022 for the IVDR), subject to the transitional provisions in EU MDR Article 120(3) and Article 110(3), respectively.
-
What is an Active Device?
‘Active device’ means any device, the operation of which depends on a source of energy other than that generated by the human body for that purpose, or by gravity, and which acts by changing the density of or converting that energy. Devices intended to transmit energy, substances or other elements between an active device and the patient, without any significant change, shall not be deemed to be active devices. Software shall also be deemed to be an active device.
Article 2.
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745 -
What’s is the classification of my device?
Advice to see documents:
- Classification Rules as per MDR Annex VIII
- MDCG 2021-24 “Guidance on classification of medical devices”
-
Where can I find he MDA/ EMDN codes?
MDA / MDN codes reflect the design and intended purpose of the device and hence are mostly relevant for the allocation of personnel involved in the review of technical documentation. In some specific cases, the NB may assign product reviewers to assess product performance and safety aspects during an audit.
Example 1: A surgical laser for refractive surgery of the eye is assigned to MDA 0302 Active
non-implantable devices utilising non-ionizing radiation and not to MDA 0309 Active nonimplantable ophthalmologic devices because, even though both codes are specific for the device, since MDA 0302 is higher in the list.
Example 2: A screw for orthopaedic surgery is assigned to MDN 1102 Non-active osteo- and orthopaedic implants and not to 1104 Non-active soft tissue and other implants, because MDN 1102 is higher in the list.European Medical Device Nomenclature (EMDN)
The European Medical Device Nomenclature (EMDN) aims at supporting the functioning of the European database on medical devices (EUDAMED). Among its various uses, it will be utilised by manufacturers for the registration of medical devices in EUDAMED, where it will be associated to each Unique Device Identifier – Device Identifier (UDI-DI). As the EMDN primarily serves regulatory purposes to support MDR and IVDR requirements, it also plays a key role in MDR/IVDR device documentation and technical documentation, sampling of technical documentation conducted by notified bodies, post-market surveillance, vigilance and post-market data analysis, etc. It is intended to support all actors in their activities under the MDR/IVDR and provides key device descriptions to patients as regards their own devices and all other devices available on the market and registered in EUDAMED.For more information:
https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2019_14_mdr_codes_en_0.pdf
https://webgate.ec.europa.eu/dyna2/emdn/ -
Am I an Economic Operator?
‘Economic operator’ means a
- manufacturer,
- an authorised representative,
- an importer,
- a distributor or
- the person referred to in Article 22(1) – systems and procedure packs and 22(3) - Any natural or legal person who sterilises systems or procedure packs.
If you are one of these, you are an economic operator.
-
What are my obligations under the MDR?
See articles of the MDR.
10 - General obligations of manufacturers,
11- Authorised representative,
13 - General obligations of importers,
14 - General obligations of distributors. -
What is the differences between MDD and MDR?
The difference between MDD and MDR is that MDD stands for Medical Device Directive, while MDR stands for Medical Device Regulation. They are both legal frameworks that regulate the medical device market in the European Union, but they have some key differences:
- MDD is a directive, meaning it is a set of rules that each EU member state has to implement in their own national legislation.
- MDR is a regulation, meaning it is directly applicable in all EU countries without the need for national legislation. This ensures a more uniform application of the rules across all EU countries, enhancing the overall consistency and transparency of the regulatory framework.
- While the MDD comprises 23 Articles and 12 annexes over 60 pages, the MDR has 123 articles and 17 annexes over 175 pages.
- MDD was adopted in 1993 and has been amended several times since then. MDR entered into force in May 2017 and will be fully applicable from May 2020, after a transition period of three years. Due to the amendment MDR replaces MDD and two other directives related to active implantable medical devices and in vitro diagnostic medical devices.
- MDR introduces several changes and enhancements compared to MDD, such as:
- Expanded definition of medical devices: The MDR broadens the definition of a medical device to clearly include products for cleaning, disinfection, or sterilization of devices.
- Increased clinical evidence requirements: The MDR requires more robust and extensive clinical data to demonstrate the safety and performance of medical devices, especially for high-risk devices. The MDR also establishes a new system of clinical evaluation consultation procedures for certain high-risk devices.
- Enhanced post-market surveillance and vigilance: The MDR strengthens the obligations of manufacturers, importers, distributors, and notified bodies to monitor the quality, safety, and performance of medical devices throughout their lifecycle. The MDR also introduces new reporting requirements and timelines for incidents and field safety corrective actions.
- Improved traceability and transparency: The MDR establishes a new system of unique device identification (UDI) for all medical devices, which will facilitate their identification and traceability throughout the supply chain. The MDR also creates a new EU database (EUDAMED) that will store and provide public access to information on medical devices, such as UDI, certificates, clinical investigations, incidents, recalls, etc.
-
What are the key changes introduced by MDR?
- The new regulation is four times longer and contains five more annexes than its predecessor, the Medical Device Directive (MDD).
- The word "safety" appears 290 times in the MDR. The MDD, by comparison, uses it only 40 times.
- Significant changes in wording used in the new law will require companies to rationalize their portfolios and perform a global impact assessment in order to implement the necessary changes to remain compliant.
- Annex I, General Safety and Performance Requirements identify new conditions that will need to be addressed for most legacy devices (CE marked under the MDD). Existing products must be re-certified in accordance with the new regulations.
- The new rules will require most companies to update clinical data, technical documentation, and labelling.
- Unique Device Identification (UDI) will be implemented to help track devices throughout the economic operator supply chain and will be required on all labels.
- While the scope of the MDD did not encompass non-medical purpose devices and AIMD, these are both included under MDR.
- The definition of medical device will be broadened to include some non-medical and cosmetic devices not previously regulated. Examples include products like non-medical contact lenses, liposuction equipment, or epilation lasers.
- Manufacturers will need to generate and provide more in-depth clinical data to prove safety and performance claims including tighter equivalency standards.
- Manufacturers will need to report all incidents, injuries, and deaths into an EU portal that will centralize relevant data so that patients have access to more safety-related information. Reporting for incidents that did not result in death or serious deterioration in health is moved to 15 days from 30 days.
- Companies undergoing transition will need to revisit core processes including the quality assurance, risk management, and post market expectations. These will require careful review, planning and updating to re-implement in compliance with new requirements.
- Reclassification of many medical devices to a higher risk class and a new classification for reusable surgical devices requiring notified body oversight.
-
What are the timelines for transition according to Regulation 2023/607?
Manufacturers with legacy devices that intend to transition to the MDR and holding Directives certificates issued from 25 May 2017, that were still valid on 26 May 2021 and that have expired before 20 March 2023 are allowed to continue placing on the market legacy devices and benefit from the extended transition timelines until 26 May 2024 if one of the following conditions is fulfilled:
- The manufacturer had signed a formal written agreement with a Notified Body for the Regulation prior to the expiry of those Directive certificates. OR
- A derogation/exemption has been granted by a Competent Authority under either Article 59(1) or Article 97(1) of the MDR before 20 March 2023.
But the manufacturer can also benefit from the longer validity of Directive certificates for legacy devices if the following conditions are met:
- No later than 26 May 2024, manufacturers have put in place an MDR compliant quality management system and have lodged a formal application with a Notified Body for MDR Conformity Assessment .
- No later than 26 September 2024, a formal agreement with a Notified Body has been signed in respect of the device covered by the expired certificate and appropriate surveillance must be guaranteed for legacy devices.
-
What is mandatory for the companies to have access to this extended period?
Extension of the transition period
This provision amends Article 120(3) MDR.- The transition period is extended from 26 May 2024 until 31 December 2027 for higher risk devices (class III and class IIb implantable devices except certain devices for which the MDR provides exemptions, given that these devices are based on well-established technologies)
- and until 31 December 2028 for medium and lower risk devices (other class IIb devices and class IIa, class Im, Is and Ir devices).
- The amendment introduces a transition period until 26 May 2026 also for class III custom-made implantable devices, which are currently not covered by Article 120(3) MDR. While manufacturers of class III implantable custom-made devices are required to comply with all applicable MDR requirements since 26 May 2021, they will now be given more time to obtain certification of their quality management system by a notified body. Also in this case, the transition period only applies if the manufacturer has lodged an application before 26 May 2024 resulting in the signing of a contract with the notified body before 26 September 2024.
In the same way as the current Article 120(3) MDR, the extended transition period applies only to ‘legacy devices’, i.e. those covered by a certificate or declaration of conformity issued under Council Directives 90/385/EEC or 93/42/EEC before 26 May 2021.
Moreover, the application of the extended transition period is subject to several cumulative conditions, which are:
- the devices must continue to comply with Directive 90/385/EEC or Directive 93/42/EEC, as applicable.
- the devices do not undergo significant changes in the design and intended purpose.
- the devices do not present an unacceptable risk to the health or safety of patients, users or other persons, or to other aspects of the protection of public health.
- no later than 26 May 2024, the manufacturer has put in place a quality management system (QMS) in accordance with Article 10(9) of the MDR. This condition aims to ensure that manufacturers gradually move towards full compliance with the MDR requirements. No specific attestation, i.e. no self-declaration nor verification of the appropriateness of the QMS by a notified body, is required at this stage. However, by submitting an application for conformity assessment to a notified body (see next condition), the manufacturer implicitly confirms that its QMS follows the MDR;
- no later than 26 May 2024, the manufacturer, or its authorised representative, has lodged a formal application in accordance with Annex VII, Section 4.3, of the MDR for conformity assessment in respect of a ‘legacy device’ covered by a Directive’s certificate or declaration of conformity, or in respect of a device intended to substitute that device under the MDR, and
- no later than 26 September 2024 the notified body and the manufacturer have signed a written agreement in accordance with Annex VII, Section 4.3, of this Regulation. This condition aims to ensure that only devices that the manufacturer intends to transition to the MDR will benefit from the extended transition period.
Reasons for the extension of period: https://health.ec.europa.eu/system/files/2023-01/mdr_proposal.pdf
-
Who does the extension of the transition period for MDR apply to?
The extension applies to ‘legacy devices’ (i.e., those covered by a certificate or declaration of conformity issued under Council Directives 90/385/EEC -Active Implantable Medical Device or 93/42/EEC – Medical Devices, before 26 May 2021).
The legacy devices covered by a certificate issued under the AIMDD or MDD remain subject to ‘appropriate surveillance’ by the notified body that issued the certificate. Alternatively, the manufacturer can agree with a notified body designated under the MDR that the latter becomes responsible for the surveillance.
At the latest by the date when the written agreement between the manufacturer and the notified body for conformity assessment in accordance with the MDR needs to be signed, that notified body would by default become responsible for the appropriate surveillance.
-
Why I must apply for a “new” MDR even if my devices are MDD certified for many years, can’t it be a simple renewal?
The MDR was issue without grand-fathering, it means all medical devices with the CE mark under the directives have to apply again for the Regulation due to all changes (explain above) (also, the Notified Bodies had to make their application for the MDR – it was not accepted a “renewal”).
-
What is a CE mark, and how does it relate to MDR?
Before a medical device can be launched in the European Economic Area (EEA), it must receive a CE mark. The CE marking indicates that the legal manufacturer has assessed the device and that it meets the General Safety and Performance Requirements under the MDR 2017/745. You need to check if your product has to be tested by a notified body. You can find this information in the relevant legislation applicable to your product. If you need to involve a notified body, the CE marking must be accompanied by the identification number of the notified body. The CE mark and the identification number can be affixed separately, as long as they appear clearly linked to each other.
You can use the Nando database to search for a notified body that can certify your product. https://webgate.ec.europa.eu/single-market-compliance-space/#/notified-bodies
If your product doesn't need to be verified by an independent body, then it is up to you to check that it complies with the technical requirements. This includes estimating and documenting the possible risks when using your product.
CE marking – obtaining the certificate, EU requirements - Your Europe (europa.eu).The legal manufacturer will verify the conformity with all relevant EU requirements and must state this in their EU Declaration of Conformity (DoC).
-
How can manufacturers ensure compliance with MDR?
The importance to understand the full range of responsibilities of medical device manufacturers under the MDR.
Article 10 MDR outlines the general requirements for all medical device manufacturers and other economic operators under the MDR.
Article 10 requires that all manufacturers must:- Ensure that their medical devices are designed and made in accordance with MDR
- Have a Risk Management system following Annex I Section 3 of the MDR
- Conduct a Clinical Evaluation of all their devices
- Maintain a record of up-to-date technical documentation
- Have access to a Person Responsible for Regulatory Compliance (PRRC)
- Have an implemented Quality Management System (QMS)
- Understanding the EU MDR
If MDR compliance has been successfully demonstrated following a suitable conformity assessment (involving a Notified Body), manufacturers are required to affix a CE-mark to their devices and must comply with requirements for using a Unique Device Identification (UDI) system. They must also ensure that procedures are in place to ensure that device production continues to be in conformity with MDR as it scales, with any changes declared to regulators, according the contract agreement with the Notified Body.
-
What are the penalties for non-compliance with MDR?
The new medical devices regulation aims to improve market transparency in Europe, product traceability, and consumer safety. Having a non-compliant device on the market exposes you to many risks.
- A financial loss
In particular, the financial loss related to non-compliance is consequential. Potential financial penalties from the authorities have to be considered here, but not only! The recall of a product, for example, can lead to significant costs. Finally, lawsuits may be initiated by consumers: the expenses linked to this type of incident are substantial. - Risks related to brand image
Company's image can quickly be affected by a non-compliant MD (Medical Device). The press quickly grasps this type of subject, and the emergence of numerous consumer associations enables consumers to exchange information more easily and quickly. All this is, of course, facilitated by digital tools and their multiplication in recent years. - Sanctions by Member States
The Member States of the European Union must notify the European Commission of the penalties and sanctions they will apply in the event of non-compliance with Regulation 2017/745. These sanctions may be financial penalties or even product withdrawals or recalls. We will come back to you towards the end of February with the specific elements.
- A financial loss
-
Is MDR only applicable to medical devices manufactured and/or put on the market in the EU?
The MDR are directly applicable to all EU Member States and therefore create a level playing field across the EU market.
Manufacturers in third countries wishing to place devices on the EU market should familiarise themselves with the rules, timelines, and obligations applicable under the Regulations before placing a medical device in the EU.Turkey had already harmonized the EU medical device directives (MDD, IVDD, AIMDD). In the meantime, the Turkish Medicines and Medical Device Agency has reconciled Turkish medical device and IVD regulations with the EU framework (MDR, IVDR). The Turkish law is also called Medical Device Regulation.
-
Is the CE mark according to MDR accepted in Great Britain (England, Wales and Scotland) and Switzerland?
Great Britain and Switzerland have their own legislation related to Medical Devices.
GB: The amended Medical Device Regulations (UK MDR 2002) https://www.legislation.gov.uk/uksi/2002/618/contents/made extend the recognition of CE marked medical devices on the Great Britain market. Medicines and Healthcare products Regulatory Agency (MHRA) also clarifies the deadlines for placing CE marked IVDs and medical devices on the Great Britain market. The timeline depends on the class of the device under the Directive/Regulation. Class I devices that are EU MDR compliant or Class A devices that are EU IVDR compliant can be marketed on the GB market until 30 June 2030. Class IIb devices can rely on the extended CE MDD Certificate in the UK until 30 June 2028 (if conditions for extension apply in the EU) and until 30 June 2030, if covered by a valid CE MDR Certificate. While, Class III devices can be marketed on the GB market until 31 December 2027 with their MDD CE Certificate (if conditions for extension apply in the EU) or until 30 June 2030, if covered by a valid MDR CE Certificate.
Northern Ireland continues to follow EU rules on medical devices and a CE marking is required for devices placed on the Northern Ireland market.Switzerland: Switzerland has recently updated its regulations on medical devices to align with the new EU Medical Device Regulations (MDR). The Medical Devices Ordinance (MedDO) https://www.fedlex.admin.ch/eli/cc/2020/552/en has been completely revised, and supplementary provisions have been added to the implementing regulations on medical devices 1. These new regulations came into effect on May 26, 2021. The Swiss authorities (Swissmedica) accept EU MDR-passed devices (devices that received the CE mark).
-
I have my medical device with the CE mark under the Directive. Do I need to apply for the MDR?
Yes, if you want to continue to sell in Europe Single Mark and Turkey (UK and Switzerland follow specific rules). Otherwise, you will not be able to sell them anymore after the expiration date of your current certificate.
-
What is the role of Notified Bodies under MDR?
A notified body is an organisation designated by an European Competent Authority to assess the conformity of certain products before being placed on the market.
These bodies carry out tasks related to conformity assessment procedures set out in the applicable legislation, when a third party is required.
Kiwa Medical has the following NoBo’s for the MDR:- Kiwa Cermet Italy (KCI) (NB0476)
- Kiwa Dare, Netherlands (NB1912)
And -for the Directive 93/42/EEC and also, branch of KCI
- Kiwa Belgelendirme Hizmetleri (NB1984)
-
Does Kiwa have the resources to do assessments for the new customers?
The Kiwa Medical Notified Bodies for the MDR are working to maintain their capacity to deliver the quotations, technical review, assessments, clinical and decision making in time to maintain their compromise with the clients.
-
How can we guarantee local speaking auditors in case it is customer requirement?
The process of having local assessors is ongoing but it will require time from the NoBo’s to qualify all the local assessors.
For some countries it will not be interested due to the labor costs.The medical device economic operators need to have their Technical Documentation in English and if, required they can pay for a translator from our assessors (to do an assessment in other EU - all of them will speak English).
Let them know that all Technical Documentation needs to be in English.
-
What is the maximum lead time for a Proposal?
Within 15 working days after the application is totally fill in.
-
What is a lead time from signed contract to Certificate?
Not less than 12 months from the moment Kiwa Medical received the technical documentation.
(In the recent EU Notified Bodies Survey on Certifications and Applications (MDR/IVDR), 25 October 2023: 45% of Notified Bodies indicated they have a 6 to 12 months lead time to issue an MDR Quality Management System (QMS) certificate.40% of Notified Bodies indicated they have a 13 to 18 months lead time to issue MDR QMS and Product Certificates. The lead times are likely to worsen as the new deadlines approach.)
-
What is the cost of a certification and a CE mark?
The costs will be detailed in our proposals and our rates are public in the web sites of the NoBo’s.
-
Is there a structure of technical documentation you recommend?
Technical documentation contains detailed information about the:
medical device, its intended use, specifications, design, manufacturing process, composition, risk management, product verification and validation and post-market activities.Requirements regarding the technical documentation are covered in Annex II and III of the MDR.
According to Annex II, technical documentation is divided into the following sections:- Device description and specifications, including variants and accessories
- Information to be supplied by the manufacturer
- Design and manufacturing information
- General safety and performance requirements
- Benefit-risk analysis and risk management
- Product verification and validation.
For more information consult Team-NB Position Paper: Best Practice Guidance for the Submission of Technical Documentation under Annex II and III of Medical Device Regulation (EU) 2017/745 https://www.team-nb.org/team-nb-position-paper-on-bpg-technical-documentation/
-
How can I manage the changes in my product according to MDR?
If your change is considered substantial then you need to contact your NoBo and manage it according to their indications.
-
Could you pre-view our technical/clinical documentation before formally lodge our application?
Not possible. As NoBo’s we can only assess the Technical Documentation under the signed agreement.
Under that agreement we can perform a Gap Analysis. -
What is the PRRC (Person Responsible for Regulatory Compliance)?
Article 15 MDR states that every manufacturer must have access to a PRRC dedicated responsibility for regulatory activities at the company, and must be allowed to undertake their duties without prejudice, even if their proper activities may result in an action that is commercially disadvantageous to the company (for example, requiring withdrawal of a device from the market on safety grounds).
Most companies will be required to employ their own responsible person.
However, the MDR does make allowances for small manufacturers who need not directly employ their own person but must have one “permanently and continually” at their disposal.
-
What is the UDI (Unique Device Identification System)?
Articles 27-31 MDR require manufacturers to ensure that their devices can be uniquely identified according to a specific, EU-wide UDI system. The MDR also requires that manufacturers keep a record of the UDI of their devices and, for higher risk devices, maintain a record of where all devices have been sold to.
-
What is the IFU – Instructions for Use?
‘Instructions for use’ means the information provided by the manufacturer to inform the user of a device's intended purpose and proper use and of any precautions to be taken.
-
Where to send my questions or application form?
All leads or direct contacts for certification and CE mark should be sent to the email address:
MEDICAL@KIWA.COM
This mail address can be accessed by the designated personnel of the Notified/Approved Bodies and your lead will be handled by Kiwa Medical according to the following internal organization:
All non-active devices: Kiwa Cermet Italy (KCI)
Active devices (devices with any type of power source):
- Netherlands, Belgium, Luxembourg, Germany, Sweden, Finland, Norway, Denmark: Kiwa Dare
- Other European countries, North America, South America, Australia, New Zealand, Africa: KCI
- Turkey, China, Korea, Pakistan, India, East Asia: Kiwa Turkey (branch of KCI)
Note1: Due to the scope of the Notified Bodies, medical devices that are under the codes MDA 0201
(active non-implantable imaging devices utilizing ionizing radiation) and MDA 0314 (active nonimplantable devices for processing and preservation of human cells, tissues or organs including in vitro fertilization and assisted reproductive technologies) will be handled by KCI.
Note2: If your lead wants a specific Notified Body, there will be no problem, and this will be handled internally by Kiwa Medical. -
Where can I see information from Kiwa Medical?
Website: Medical devices - Kiwa
LinkedIn: https://www.linkedin.com/showcase/kiwa-medical/ -
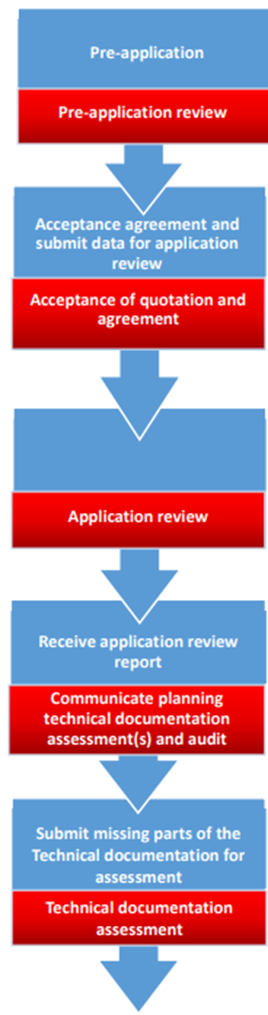
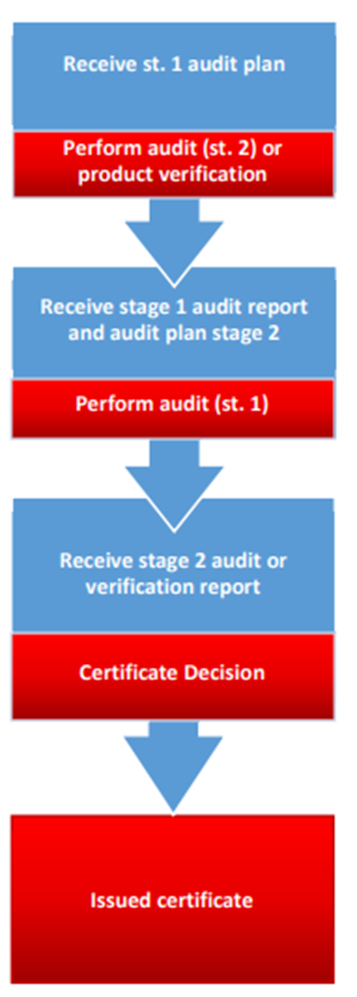
Do you have a synthetic guideline to better understand the process and certification flow?